https://doi.org/10.35381/s.v.v8i1.3819
Síndrome de ictiosis arlequín hereditaria: Desafío diagnóstico prenatal
Hereditary harlequin ichthyosis syndrome: Prenatal diagnostic challenge
Juan Alberto Viteri-Rodríguez
Universidad Regional Autónoma de Los Andes, Ambato, Tungurahua, Ecuador
https://orcid.org/0000-0002-2463-7036
Emily Valeria Arcos-Aldaz
ma.emilyvaa02@uniandes.edu.ec,
Universidad Regional Autónoma de Los Andes, Ambato, Tungurahua, Ecuador
https://orcid.org/0000-0002-7627-648X
Natali Mishell Duarte-Cortez
ma.natalimdc35@uniandes.edu.ec
Universidad Regional Autónoma de Los Andes, Ambato, Tungurahua, Ecuador
https://orcid.org/0000-0002-1336-8846
Revisado: 10 de diciembre 2023
Aprobado: 15 de enero 2024
Publicado: 01 de febrero 2024
RESUMEN
Objetivo: analizar el síndrome de Ictiosis Arlequín hereditaria: desafío diagnóstico prenatal. Método: Descriptiva documental. Conclusión: La HI, también conocida como queratosis difusa fetal, es la forma más grave y devastadora de las ictiosis congénitas autosómicas recesivas. Se debe a mutaciones en el transportador ABCA12, un transportador de la membrana celular asociado al transporte de lípidos. Los fetos portadores de esta mutación producen una secreción defectuosa de lípidos en los queratinocitos epidérmicos, lo que provoca una pérdida de la barrera lipídica de la piel y el desarrollo de arlequín. La incidencia de la enfermedad es muy baja y no existe un tratamiento efectivo. La probabilidad de mortalidad de los niños con IH es alta, su pronóstico es malo y la mayoría de los neonatos mueren poco después del parto debido a infecciones, pérdida de calor, deshidratación, alteraciones electrolíticas o dificultad respiratoria.
Descriptores: Estructuras embrionarias; atención prenatal; diagnóstico prenatal. (Fuente: DeCS).
ABSTRACT
Objective: to analyze hereditary harlequin ichthyosis syndrome: prenatal diagnostic challenge. Method: Descriptive documentary. Conclusion: HI, also known as diffuse fetal keratosis, is the most severe and devastating form of autosomal recessive congenital ichthyosis. It is due to mutations in the ABCA12 transporter, a cell membrane transporter associated with lipid transport. Fetuses carrying this mutation produce defective lipid secretion in epidermal keratinocytes, leading to a loss of the skin lipid barrier and the development of harlequin. The incidence of the disease is very low and there is no effective treatment. The probability of mortality of infants with HI is high, their prognosis is poor and most neonates die shortly after delivery due to infections, heat loss, dehydration, electrolyte disturbances or respiratory distress.
Descriptors: Embryonic structures; prenatal care; prenatal diagnosis. (Source: DeCS).
INTRODUCCIÓN
La ictiosis congénita autosómica recesiva (ARCI) presenta una variedad de fenotipos, incluida la devastadora enfermedad de la piel, ictiosis arlequín (HI) y está asociado con la mutación del gen ABCA12 que se encuentra en el cromosoma 2q33-q35, por lo que da lugar a la noción de que la consanguinidad entre los padres tiene un papel que desempeñar en su etiología. 1 2 3 4 5
El diagnóstico prenatal de HI consiste en un examen de ultrasonido para las anomalías morfológicas típicas, ya sea una biopsia de vellosidades coriónicas o una muestra de líquido amniótico para la prueba de mutaciones patogénicas ABCA12.67 Un diagnóstico prenatal confirmado es fundamental para el manejo perinatal y posnatal, así como para el asesoramiento genético. Es poco probable que se justifique el diagnóstico prenatal, especialmente cuando la pareja no tiene antecedentes de la enfermedad. 8 9
Se tiene por objetivo analizar el síndrome de Ictiosis Arlequín hereditaria como desafío de diagnóstico prenatal.
MÉTODO
Descriptiva documental.
Se revisaron 15 articulos científicos publicados en PubMed.
Se analizaron mediante analítica documental.
RESULTADOS
La HI es un trastorno grave pero raro que es mortal para los recién nacidos. Para hacer frente a este desafío médico único y aumentar las tasas de supervivencia, es esencial un equipo multidisciplinario de médicos, entre otros expertos. Los investigadores ya se han centrado en el aspecto genético de la enfermedad. 10 11 12
A pesar del conocimiento del gen responsable, las mutaciones presentan varias disparidades entre cada caso, lo que lleva a una variedad de síntomas y afecta las tasas de supervivencia. La comprensión total de las mutaciones del gen ABCA12 es vital para controlar el trastorno. Los mecanismos fisiopatológicos responsables de la manifestación de la enfermedad están asociados con las mutaciones creadas en el genoma y, por lo tanto, los investigadores sugieren que este conocimiento aumentará las tasas de supervivencia después del nacimiento.
Durante el período perinatal, el paciente portador de una enfermedad rara requiere de especialistas lo que involucra a obstetras, genetistas, radiólogos, cirujanos infantiles, pediatras neonatólogos y psicólogos, entre otros. Sin este enfoque, el diagnóstico y manejo se hace extremadamente difícil. La evaluación prenatal es la primera aproximación a un defecto fetal, pero se debe prestar atención a un examen sistemático en el tercer trimestre del embarazo según las características clínicas de la enfermedad (Figura 1). Es en esta etapa en la que se aproxima el diagnóstico y etiología más probable de la enfermedad, para diseñar un plan de seguimiento y modo de nacimiento
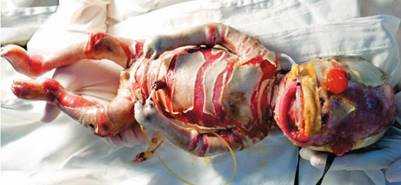
Figura 1. Recién nacido con encajamiento grueso hiperqueratósico con fisuras, al presentarse en el servicio de urgencias.
Fuente: 13
Para confirmar el diagnóstico y daño génico en pacientes con ictiosis hereditarias, se ha utilizado desde hace más de 30 años el análisis de ADN por el método de secuenciación de Sanger, lo que ha representado desafíos por su alto costo y el tiempo necesario para desarrollar la prueba. Actualmente, se han desarrollado otros métodos de diagnóstico molecular con resultados fiables, como la biopsia de piel, que pueden tener más repercusiones, especialmente en este tipo de pacientes. También es posible el diagnóstico prenatal, la identificación de la mutación del gen con análisis de ADN mediante muestreo de células de líquido amniótico o vellosidades coriónicas en etapas más tempranas del embarazo o el diagnóstico mediante ultrasonido 3D/4D, ya que estos métodos pueden observar signos sugestivos de ictiosis hereditaria
Las pruebas de detección prenatales para HI involucran procedimientos invasivos, como biopsias de piel fetal guiadas por ecografía que revelan características ultraestructurales clave, como gránulos lamelares multivesiculares en fetos afectados a las 20-24 semanas de gestación. Se han descrito cambios morfológicos similares en las células del líquido amniótico a las 17 semanas de gestación.
Otros métodos que se han utilizado en los últimos años para el diagnóstico prenatal de HI son la ecografía tridimensional (Figura 2) y tetradimensional en tiempo real.
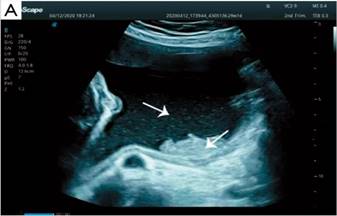
Figura 2. Ecografía tridimensional. Partículas flotantes en el líquido amniótico (flechas). prenatal con genotipado rápido de SNP-array en sangre del cordón umbilical.
Fuente: 14
Un beneficio importante de los hallazgos actuales es que ahora debería ser posible el análisis basado en el ADN, lo que facilita las pruebas prenatales más tempranas (es decir, en el primer trimestre) que son más confiables y concluyentes que las otras técnicas de detección prenatal no basadas en el ADN. A continuación, los diferentes tipos de diagnósticos en el área prenatal que determinan la presencia de este síndrome.
La aplicación tecnológica de genotipado rápido de SNP-array identifica mediante el mapeo de homocigosidad, las mutaciones ABCA12 como la principal causa genética subyacente de la devastadora enfermedad de la piel HI. La asociación de ABCA12 encargado del transporte de lípidos, la formación de barreras y la diferenciación de queratinocitos, las mutaciones con HI facilitarán el diagnóstico de ADN prenatal de este trastorno potencialmente mortal, así como la posibilidad de realizar pruebas genéticas previas a la implantación. La mayor comprensión de este gen puede conducir al desarrollo de una terapia dirigida para disminuir la carga de esta enfermedad tanto para los niños sobrevivientes como para sus padres. 15
CONCLUSIONES
La HI, también conocida como queratosis difusa fetal, es la forma más grave y devastadora de las ictiosis congénitas autosómicas recesivas. Se debe a mutaciones en el transportador ABCA12, un transportador de la membrana celular asociado al transporte de lípidos. Los fetos portadores de esta mutación producen una secreción defectuosa de lípidos en los queratinocitos epidérmicos, lo que provoca una pérdida de la barrera lipídica de la piel y el desarrollo de arlequín. La incidencia de la enfermedad es muy baja y no existe un tratamiento efectivo. La probabilidad de mortalidad de los niños con IH es alta, su pronóstico es malo y la mayoría de los neonatos mueren poco después del parto debido a infecciones, pérdida de calor, deshidratación, alteraciones electrolíticas o dificultad respiratoria.
El riesgo de recurrencia en el embarazo es del 25%, por lo que el diagnóstico prenatal es importante. En pacientes con antecedentes familiares realizar fetoscopia con biopsia de piel y examen ultraestructural del canal capilar y de las células fútiles amnióticas puede ser útil. En el caso de las personas sin antecedentes familiares, muchos casos se pasan por alto, y se debe principalmente a que la queratinización no comienza hasta las 22 a 24 semanas de gestación en el feto normal. Tras el descubrimiento de la mutación ABCA12, el diagnóstico puede confirmarse de vellosidades coriónicas o muestras de líquido amniótico, pero en pacientes sin antecedentes familiares, el gen diana no se de forma rutinaria.
La
mayoría de los rasgos característicos de la ecografía sólo se ponen de
manifiesto más adelante en el embarazo cuando el desarrollo restringido de la
piel se convierte en una limitación para el crecimiento y los movimientos del
feto. Por lo que el diagnóstico de HI en el período prenatal es![]() importante
con el examen ultrasonografía y el cribado de la mutación del Gen ABC 12, en el
caso de que se quiera dar la interrupción del embarazo será posible, o en el
caso que decidan continuar con el embarazo el asesoramiento genético es muy
importante para las familias con matrimonios consanguíneos que aportan
antecedentes de niños con HI para esta grave enfermedad que no se puede
prevenir, pero un adecuado diagnóstico temprano puede garantizar medidas de
soporte inicial para prevenir complicaciones y ofrecer un manejo
multidisciplinario a estos pacientes.
importante
con el examen ultrasonografía y el cribado de la mutación del Gen ABC 12, en el
caso de que se quiera dar la interrupción del embarazo será posible, o en el
caso que decidan continuar con el embarazo el asesoramiento genético es muy
importante para las familias con matrimonios consanguíneos que aportan
antecedentes de niños con HI para esta grave enfermedad que no se puede
prevenir, pero un adecuado diagnóstico temprano puede garantizar medidas de
soporte inicial para prevenir complicaciones y ofrecer un manejo
multidisciplinario a estos pacientes.
Los autores declaran que no tienen conflicto de interés en la publicación de este artículo.
FINANCIAMIENTO
Autofinanciado.
AGRADECIMIENTO
A todos los actores sociales involucrados en el desarrollo de la investigación.
REFERENCIAS
1. Mohamad J, Samuelov L, Assaf S, et al. Autosomal recessive congenital ichthyosis caused by a pathogenic missense variant in CLDN1. Am J Med Genet A. 2022;188(10):2879-2887. https://doi.org/10.1002/ajmg.a.62924
2. Zaouak A, Chamli A, Ben Mansour N, Jouini W, Fenniche S, Hammami H. Nail involvement in autosomal recessive congenital ichthyosis. Clin Dermatol. 2022;40(4):388-394. https://doi.org/10.1016/j.clindermatol.2022.02.012
3. Richard G. Autosomal Recessive Congenital Ichthyosis. In: Adam MP, Feldman J, Mirzaa GM, et al., eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; January 10, 2001.
4. Youssef G, Ono M, Brown SJ, et al. Identifying a hyperkeratosis signature in autosomal recessive congenital ichthyosis: Mdm2 inhibition prevents hyperkeratosis in a rat ARCI model. J Invest Dermatol. 2014;134(3):858-861. https://doi.org/10.1038/jid.2013.374
5. Sathishkumar D, Peter D, Pulimood S, et al. Bathing Suit Variant of Autosomal Recessive Congenital Ichthyosis (ARCI) in Two Indian Patients. Case Rep Dermatol Med. 2018;2018:3140473. https://doi.org/10.1155/2018/3140473
6. Hotz A, Kopp J, Bourrat E, et al. Mutational Spectrum of the ABCA12 Gene and Genotype-Phenotype Correlation in a Cohort of 64 Patients with Autosomal Recessive Congenital Ichthyosis. Genes (Basel). 2023;14(3):717. https://doi.org/10.3390/genes14030717
7. Fioretti T, Auricchio L, Piccirillo A, et al. Multi-Gene Next-Generation Sequencing for Molecular Diagnosis of Autosomal Recessive Congenital Ichthyosis: A Genotype-Phenotype Study of Four Italian Patients. Diagnostics (Basel). 2020;10(12):995. https://doi.org/10.3390/diagnostics10120995
8. Nishimura G, Handa A, Miyazaki O, et al. Prenatal diagnosis of bone dysplasias. Br J Radiol. 2023;96(1147):20221025. https://doi.org/10.1259/bjr.20221025
9. Meller CH, Grinenco S, Aiello H, et al. Congenital heart disease, prenatal diagnosis and management. Cardiopatías congénitas, diagnóstico y manejo prenatal. Arch Argent Pediatr. 2020;118(2):e149-e161. https://doi.org/10.5546/aap.2020.eng.e149
10. Mohamad J, Nanda A, Pavlovsky M, et al. Phenotypic suppression of acral peeling skin syndrome in a patient with autosomal recessive congenital ichthyosis. Exp Dermatol. 2020;29(8):742-748. https://doi.org/10.1111/exd.14140
11. Kiener S, Wiener DJ, Hopke K, et al. ABHD5 frameshift deletion in Golden Retrievers with ichthyosis. G3 (Bethesda). 2022;12(2):jkab397. https://doi.org/10.1093/g3journal/jkab397
12. Zaouak A, Abdessalem G, Chamli A, et al. Coexistence of Ichthyosis, Yellowish Keratoderma, and Neurologic Manifestations: Think About Sjogren-Larsson Syndrome. Skinmed. 2023;21(6):457-458.
13. Tsivilika M, Kavvadas D, Karachrysafi S, Sioga A, Papamitsou T. Management of Harlequin Ichthyosis: A Brief Review of the Recent Literature. Children (Basel). 2022;9(6):893. Published 2022 Jun 15. https://doi.org/10.3390/children9060893
14. Zhou XJ, Lin YJ, Chen XW, Zheng JH, Zhou YJ. Prenatal diagnosis of harlequin ichthyosis by ultrasonography: a case report. Ann Transl Med. 2021;9(2):183. https://doi.org/10.21037/atm-20-8223
15. Jian W, Du QT, Lai ZF, et al. Prenatal diagnose of a fetus with Harlequin ichthyosis in a Chinese family. Taiwan J Obstet Gynecol. 2018;57(3):452-455. https://doi.org/10.1016/j.tjog.2018.04.023
©2024 por los autores. Este artículo es de acceso abierto y distribuido según los términos y condiciones de la licencia Creative Commons Atribución-NoComercial-CompartirIgual 4.0 Internacional (CC BY-NC-SA 4.0) (https://creativecommons.org/licenses/by-nc-sa/4.0/).